


Looking Forward to EU Regulation (EU) No 536/2014
Contributed Commentary by Torsten Staller, KCR
January 26, 2022 | We are facing one of the most fundamental changes in the regulatory landscape for clinical trials in Europe. It is both historic and exciting.
The EU Clinical Trials Register will be implemented on 31 January 2022, with the launch of the harmonized Clinical Trials Information System (CTIS) in all EU and EEA Member States and will repeal the EU Clinical Trial Directive (EC) No. 2001/20/EC and its interpretation in national legislation with a 3-year transition period. From the moment the CTIS goes live, Member States will have to work with the portal. Sponsors have 1 year to submit new studies under the CT Directive 2001/20/EC, but from 31-January 2023 all new studies must be submitted under the Clinical Trials Regulation (EU) No 536/2014via the CTIS. By 31 January 2025, all ongoing studies must be submitted via CTIS.

How to Get Started: Registration for CTIS
Each sponsor must register with CTIS if they do not already have a user account registered with EMA Account Management. If a user is already using EMA services, such as Eudralink, SPOR, EudraCT or IRIS, sponsor already has an EMA account and does not need to register again. The EMA account (including Organisation Management Service (OMS) registration) is a prerequisite for registration in CTIS, the first step being to register a High Level Administrator.
What are the benefits of the EU CTR?
Until now, individual Member States of the EU and the EEA had more or less divergent interpretations of the Directive. Now, the legal provisions and the processes around clinical trials will be harmonized.
While this applies to Part 1 of the submission documents, the EU would not be the EU if there were not country-specific interpretations of Part 2. Part 2 is broadly harmonized, but leaves room for each Member State to determine its own conditions regarding country specific documents required and assessment processes (involvement of national bodies in the assessment of Part 2) while staying harmonized to timelines of part I.
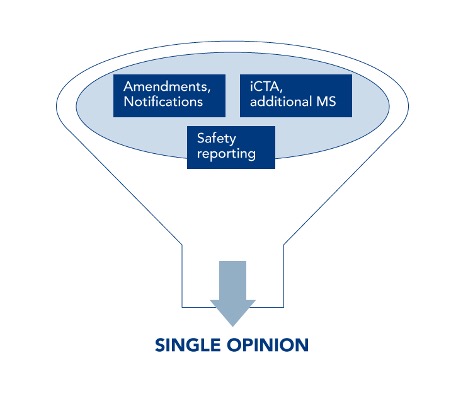
Common and legally binding is the submission via the Single Entry Point of the CTIS. This is where all submissions, deficiencies and the issuance of the Single Opinion are managed: initial submission, amendments, notifications, interim reports, End Of Trial notification and Clinical Study Report upload, as well as safety reporting and adding new countries to an already approved protocol.
The main advantages of the EU CTR are that it establishes high standards for security, transparency and information clarity/completeness and that strict deadlines are now anchored in law, which requires all parties involved to organize the internal processes well while providing reliable planning security.
What We Need to Know: Good Preparation and Expectations
The key to a good in-house implementation of the EU CTR and the related processes is a complete understanding of the portal management, as it enables the alignment of the processes that are essential to meet the legal deadlines (12 days to respond to any scientific/regulatory deficiency) and to use the full power of the EU CTR. The best possible Trainings are provided by the EMA, alongside the Clinical Trial Information System (CTIS) - Sponsor Handbook, on their webpage. Keep yourself ahead of the game and register for the CTIS Highlights Newsletter.
While there’s no reason to be concerned about the interpretation of the CTR, some adjustments to the system, especially at the beginning, are to be expected and can be solved with a professional approach. The EMA offers limited access to a CTIS sandbox for testing as announced by EMA on 19th October. Registration period for the sandbox expired on 31-Oct-2021.
So when CTIS comes into effect on 31 January 2022, and with it the implementation of the EU CTR, it will be exciting for everyone, no matter how well trained and prepared we are.
Torsten Staller is the Director of Regulatory Affairs at KCR, an international clinical development solutions provider for the biotechnology and pharmaceutical industries. He has 10+ years of experience in global management roles, providing regulatory advisements to international clinical research organizations. At KCR, Mr. Staller oversees the complete design and execution of all KCR’s global and local regulatory processes—ensuring total compliance and excellence for all KCR teams and affiliates. He holds multiple graduate degrees in the life sciences. He can be reached at Torsten.Staller@kcrcro.com.


